Next-generation sequencing has been applied to identify genomic variations possibly associated to many diseases. The next challenge consists in identifying the role of those mutations.
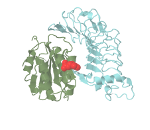
Mutations located in protein interaction interfaces are for instance often associated with loss-of-function or gain-of-function. The identification of such residues requires the integration of disseminated biological data and bioinformatics tool. Fortunately, the possibility to extend the existing genome browsers allows us to integrate complex frameworks and make them available through already widely adopted tools. We have developed an extension for the Integrated Genome Browser, a widely used tool for the visualization and analyzes of genomic data, which maps and visualizes genomic regions in molecular interactions structures (protein-protein, protein-DNA, protein-RNA and protein-small molecule interactions) and allows the researcher to make educated guesses about the functional impact of somatic mutations.
The MI Bundle is a plugin for the IGB. The plugin, help, video and tutorial are available at http://cru.genomics.iit.it/igbmibundle/.
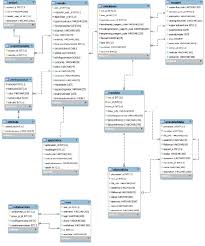
SMITH is a web application with a MySQL server at the backend. Wet-lab scientists of the Centre for Genomic Science and database experts from the Politecnico of Milan in the context of a Genomic Data Model Project developed SMITH. The data base schema stores all the information of an NGS experiment, including the descriptions of all protocols and algorithms used in the process. Notably, an attribute-value table allows associating an unconstrained textual description to each sample and all the data produced afterwards. This method permits the creation of metadata that can be used to search the database for specific files as well as for statistical analyses.
SMITH runs automatically and limits direct human interaction mainly to administrative tasks. SMITH data-delivery procedures were standardized making it easier for biologists and analysts to navigate the data. Automation also helps saving time. The workflows are available through an API provided by the workflow management system. The parameters and input data are passed to the workflow engine that performs de-multiplexing, quality control, alignments, etc.
SMITH standardizes, automates, and speeds up sequencing workflows. Annotation of data with key-value pairs facilitates meta-analysis. SMITH is available at http://cru.genomics.iit.it/smith/
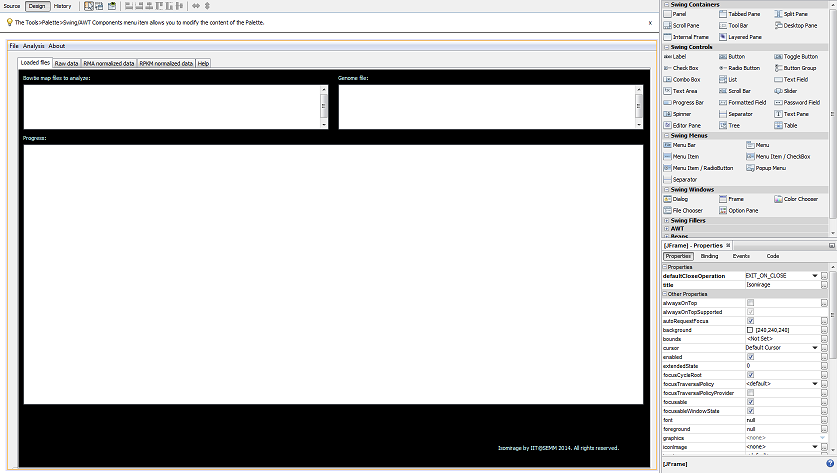
Isomirage is a desktop application that counts the number of occurrences of small RNA molecules in a Bowtie .map alignment file.As more small RNA sequencing libraries are becoming available, it clearly emerges that microRNAs (miRNAs) are highly heterogeneous both in length and sequence. In comparison to canonical miRNAs, miRNA isoforms (termed as “isomiRs”) might exhibit different biological properties, such as a different target repertoire, or enhanced/reduced stability. Nonetheless, this layer of information has remained largely unexplored due to the scarcity of small RNA NGS-datasets and the absence of proper analytical tools. Here, we present a workflow for the characterization and analysis of miRNAs and their variants in next-generation sequencing datasets. IsomiRs can originate from an alternative dicing event (“templated” forms) or from the addition of nucleotides through an enzymatic activity or target-dependent mechanisms (“non-templated” forms). Our pipeline allows distinguishing canonical miRNAs from templated and non-templated isomiRs by alignment to a custom database, which comprises all possible 3′-, 5′-, and trimmed variants. Functionally equivalent isomiRs can be grouped together according to the type of modification (e.g., uridylation, adenylation, trimming …) to assess which miRNAs are more intensively modified in a given biological context. When applied to the analysis of primary epithelial breast cancer cells, our methodology provided a 40% increase in the number of detected miRNA species and allowed to easily identify and classify more than 1000 variants. Most modifications were compatible with templated IsomiRs, as a consequence of imprecise Drosha or Dicer cleavage. However, some non-templated variants were consistently found either in the normal or in the cancer cells, with the 3′-end adenylation and uridylation as the most frequent events, suggesting that miRNA post-transcriptional modification frequently occurs. In conclusion, our analytical tool permits the deconvolution of miRNA heterogeneity and could be used to explore the functional role of miRNA isoforms. IsomiRage is available at http://cru.genomics.iit.it/Isomirage/.
Modern genomic technologies produce large amounts of data that can be mapped to specific regions in the genome. Among the first steps in interpreting the results is annotation of genomic regions with known features such as genes, promoters, CpG islands etc. Several tools have been published to perform this task. However, using these tools often requires a significant amount of bioinformatics skills and/or downloading and installing dedicated software. AnnotateGenomicRegions is a web application that accepts genomic regions as input and outputs a selection of overlapping and/or neighboring genome annotations. AnnotateGenomicRegion is accessible online on a public server or can be installed locally. The most frequently used annotations and genomes are available while custom annotations may be added by the user.
We are currently migrating some of our servers, including AnnotateGenomicRegions, which is not available for the moment.
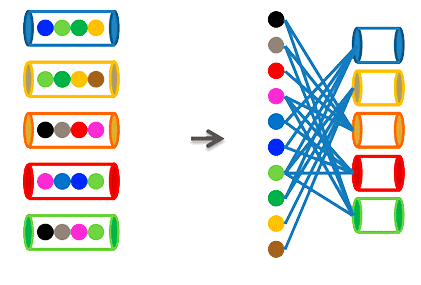
Next-generation sequencing has recently been applied to the sequencing of cancer genomes. Cancer genomes are known to be unstable and to accumulate varying numbers of somatic rearrangements and mutations. The challenge is to distinguish events that are relevant for the onset and the progression of the disease from simple bystander events. Large international consortia have formed with the aim of providing cancer genome sequencing data to the scientific community. It is hoped that the analysis of these data in a community effort is capable of providing crucial answers fast and efficiently. In order to contribute to this goal, we set out to analyze data provided by the TCGA. TCGA (The Cancer Genome Atlas) is an NIH administered effort involving more than 150 researchers from dozens of US institutions with the aim of providing biomolecular data on thousands of cancer samples from 20 different cancer types.
Cancer samples can be classified based on somatic mutation spectra using bipartite graph models. Bipartite graph models have the invaluable advantage of being capable of analyzing the data set as a whole instead of analyzing mutations in isolation for their association with a given type of tumor. The Successful classification of cancer samples permits the identification of classifier mutations who are excellent candidates for being driver mutations instead of being bystanders. Future studies are needed to validate the status of these mutations as classifiers using many more samples and to identify their molecular consequences, which may lead to novel targets of therapeutic intervention.

TCGA (The Cancer Genome Atlas) is an NIH administered effort involving more than 150 researchers from dozens of US institutions with the aim of providing biomolecular data on thousands of cancer samples from 20 different cancer types. The TCGA Annotation Browser allows for searching annotations related to the samples provided by the TCGA and is available at https://cru.genomics.iit.it/TcgaAnnotationBrowser/.